Para abordar los procesos de neurodegeneración acontecidos
en el ser humano, como en la enfermedad de Huntington, son de gran utilidad y
apoyo los modelos experimentales, tanto los que usan animales transgénicos como
los inducidos por neurotóxicos, a pesar del salto filogenético que implica su
uso.
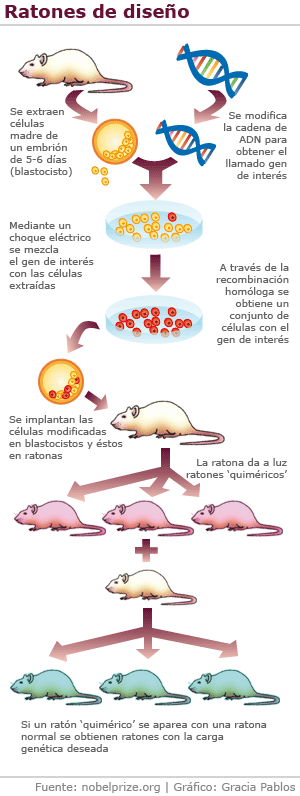 Los modelos transgénicos desarrollan cuadros con
gran similitud fenotípica, bioquímica y genética al ser humano, y muestran no
sólo los cambios conductuales, tróficos y bioquímicos característicos, sino
también las alteraciones genéticas implicadas y, en consecuencia, los depósitos
de la proteína mHtt. En la actualidad, se dispone de una gran variedad de
modelos genéticamente manipulados (transgénicos):
Los modelos transgénicos desarrollan cuadros con
gran similitud fenotípica, bioquímica y genética al ser humano, y muestran no
sólo los cambios conductuales, tróficos y bioquímicos característicos, sino
también las alteraciones genéticas implicadas y, en consecuencia, los depósitos
de la proteína mHtt. En la actualidad, se dispone de una gran variedad de
modelos genéticamente manipulados (transgénicos):
–Transgénicos propiamente denominados: aquéllos que
tienen incorporado en su genoma un fragmento de ADN exógeno con un número de
copias en tándem y al azar (por ejemplo, YAC72, YAC128, R6/, R6/2). El R6/2 es
uno de los más utilizados, ya que manifiesta una forma muy agresiva y similar a
la variante juvenil.
–Knock-in: aquéllos en los que se ha sustituido un
gen normal para la proteína Htt, insertándose una secuencia codificante o el
gen completo para la mHtt en la localización exacta dentro del brazo corto del
cromosoma 4, lo que le va a conferir otras propiedades determinadas (por
ejemplo, CAG140).
–Knock-out: inactivación de los dos alelos de un gen
(por ejemplo, Htt). Según nuestro conocimiento, estos modelos en roedores no
muestran algunas de las alteraciones cerebrales (inclusiones nucleares y
agregados de neuropilo) y modificaciones de la conducta (distonía y corea) que
padecen los pacientes con esta enfermedad. Por ello, se han desarrollado
modelos de primates no humanos transgénicos, utilizando al macaco rhesus, que
adicionalmente reproduce modificaciones cerebrales y conductuales similares a
las del ser humano (inclusiones nucleares, agregados de neuropilo, distonía y
corea).
 Por su parte, los modelos inducidos mediante agentes
neurotóxicos presentan ventajas como ser más económicos y más fácilmente
manipulables, y reproducir cambios bioquímicos, moleculares y conductuales
similares a la enfermedad estudiada. Al igual que los modelos transgénicos,
también presentan algún inconveniente o limitación, como sería, principalmente,
el no mostrar el defecto o la alteración genética. En el caso específico de los
agentes neurotóxicos reproductores de un modelo similar a la enfermedad de
Huntington (ácido quinolínico, ácido 3-nitropropiónico), no manifiestan la
expansión del triplete CAG y, por lo tanto, los depósitos de proteína mHtt. En
el caso concreto de la enfermedad de Huntington, el modelo inducido por el
ácido 3-nitropropiónico reproduce con fidelidad cambios conductuales y del
comportamiento, así como bioquímico-moleculares y celulares similares a los
acontecidos en esta enfermedad, excepto los depósitos citoplasmáticos e
intranucleares de mHtt.
Por su parte, los modelos inducidos mediante agentes
neurotóxicos presentan ventajas como ser más económicos y más fácilmente
manipulables, y reproducir cambios bioquímicos, moleculares y conductuales
similares a la enfermedad estudiada. Al igual que los modelos transgénicos,
también presentan algún inconveniente o limitación, como sería, principalmente,
el no mostrar el defecto o la alteración genética. En el caso específico de los
agentes neurotóxicos reproductores de un modelo similar a la enfermedad de
Huntington (ácido quinolínico, ácido 3-nitropropiónico), no manifiestan la
expansión del triplete CAG y, por lo tanto, los depósitos de proteína mHtt. En
el caso concreto de la enfermedad de Huntington, el modelo inducido por el
ácido 3-nitropropiónico reproduce con fidelidad cambios conductuales y del
comportamiento, así como bioquímico-moleculares y celulares similares a los
acontecidos en esta enfermedad, excepto los depósitos citoplasmáticos e
intranucleares de mHtt.

No hay comentarios:
Publicar un comentario